近期,张运院士研究团队在心血管病基础研究领域取得了一系列重大进展,在《循环研究》(Circulation Research)《细胞死亡和分化》(Cell Death and Differentiation)《治疗诊断学》(Theranostics)等国际期刊连续发表多篇高水平研究论文,受到国际学术界的高度关注。
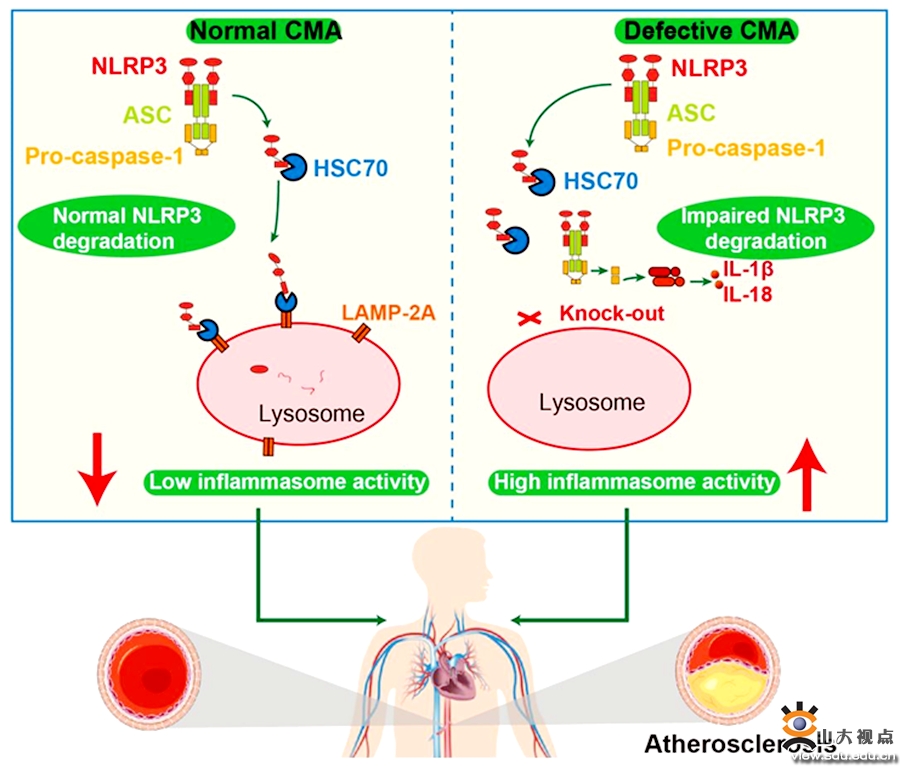
动脉粥样硬化性心血管疾病已成为我国人口死亡的首要病因。大量基础和临床研究证明,动脉粥样硬化(AS)是一种慢性炎症性疾病,2017年发表的CANTOS临床试验将IL-1β确立为AS抗炎治疗的关键靶点,使用IL-1β的单克隆抗体治疗心肌梗死后患者取得了成功。NLRP3炎性小体是IL-1β的上游调控节点,NLRP3炎性小体活化可促进 IL-1β的产生,在AS炎症反应中起关键作用。NLRP3炎性小体的降解可负性调控NLRP3炎性小体的活化,深入探讨 NLRP3炎性小体的负性调控机制,对于降低 IL-1β水平和干预AS的发生和发展具有重要意义。分 子 伴 侣 介 导 的 自 噬 (CMA)可针对性地介导细胞代谢过程中的一些关键蛋白质的降解,因而是一种特异和高效的蛋白质降解系统,但CMA能否调控NLRP3炎性小体的降解并在AS进程中发挥作用尚不明了。张运院士领导的课题组在ApoE-/-小鼠中建立了不同病程的AS模型,发现随着AS斑块的不断进展,CMA的标志物LAMP-2A表达量逐渐减少,在人体尸检标本中观察到同样的现象。课题组构建了巨噬细胞 LAMP-2A基因特异性敲除小鼠以阻断CMA的功能,结果显示,与同窝CMA功能正常且病程相同的小鼠相比,CMA缺陷小鼠的主动脉弓及分支处斑块数量明显增多,主动脉根部的斑块面积明显增大,表明CMA缺陷可促进AS斑块的进展。体外实验证明,巨噬细胞CMA缺失可导致NLRP3炎性小体的激活,进而活化半胱天冬酶 1,后者切割IL-1β和IL-18并使其活化,反之,将LAMP-2A转入CMA缺失的巨噬细胞可明显逆转NLRP3炎性小体的活化程度。课题组在体外和体内实验进一步证实,NLRP3蛋白是通过CMA途径降解的,CMA缺陷导致NLRP3蛋白降解受阻,这是CMA调控NLRP3炎性小体活化的重要机制。该研究首次发现了CMA在AS发病机制中的作用,证明巨噬细胞CMA功能缺陷可增强 NLRP3炎性小体所介导的炎症反应,从而加速AS进展,上调CMA功能有望成为治疗AS、肥胖、代谢紊乱综合征等慢性炎症性疾病的新途径。
该研究发表于美国心脏学会主办的心血管基础研究领域的国际杂志《循环研究》(Circulation Research,中科院一区,最新影响因子17.367),论文的第一作者是山东大学齐鲁医院心内科博士后乔磊和博士生马静,山东大学齐鲁医院张运院士和陈文强教授为该论文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。
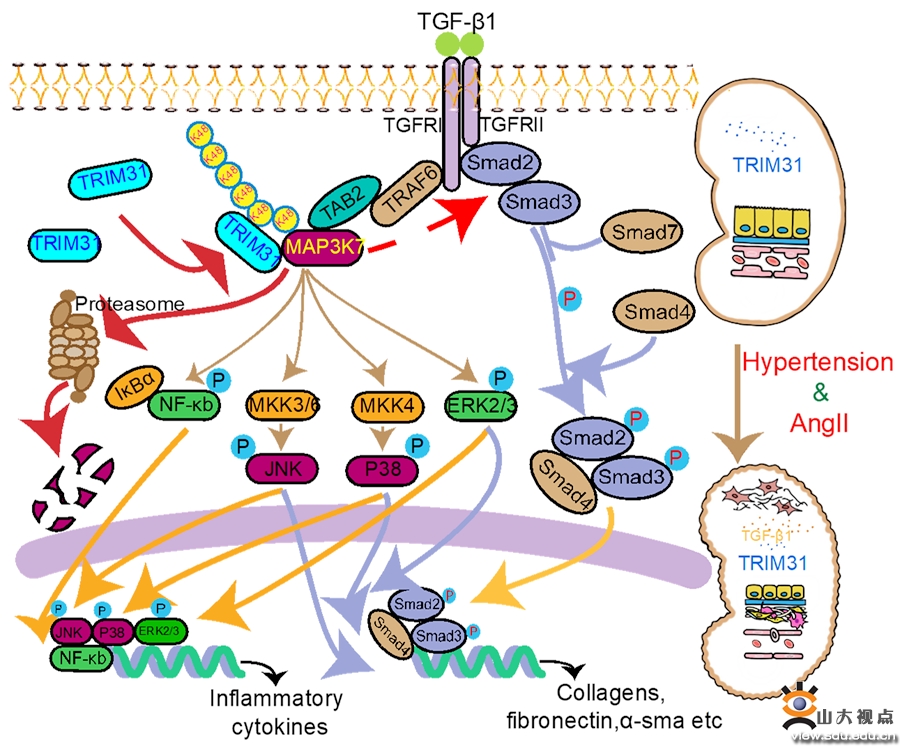
高血压肾病(HRD)是由原发性高血压导致的肾脏结构和功能的损害,主要表现为蛋白尿增多、良性肾小球硬化、肾脏间质纤维化以及炎症细胞浸润,但其发病机制尚不明了。泛素化修饰是一种重要的蛋白质翻译后修饰,在多种疾病进展中发挥着关键作用。TRIM31属于TRIM家族的一员,是一种重要的E3泛素连接酶。以往研究表明,TRIM31在先天免疫应答、NLRP3炎症小体的激活、肠道菌群的组成以及肠道自噬中发挥着关键调控作用,TRIM31是否可通过其E3泛素连接酶功能影响HRD的疾病进展尚无报道。课题组在HRD患者的肾活检标本中发现,随着患者肾脏损伤和纤维化的进展,TRIM31的表达明显下调, 在AngII输注所致HRD小鼠模型的肾脏尤其是肾小管中,TRIM31的表达同样明显下调,提示HRD的进展可能与TRIM31在肾脏中的表达下调有关。为了探索TRIM31在HRD中的作用,课题组构建了TRIM31基因敲除小鼠(TRIM31-/-),在此组小鼠和同窝野生型小鼠中分别泵入AngII持续6周以构建HRD小鼠模型。结果显示,AngII 诱导的 HRD 小鼠肾功能明显损伤,TRIM31 基因缺失进一步加剧了AngII引起的小鼠肾功能损伤、肾脏滤过屏障损害、肾纤维化和炎症反应。为了进一步验证TRIM31与AngII诱导的HRD之间的因果关系,课题组通过AAV9-TRIM31病毒注射上调小鼠肾脏中TRIM31的表达,同样泵入AngII 持续6周以构建HRD小鼠模型,发现TRIM31过表达可缓解AngII诱导的小鼠肾损伤、纤维化和炎症反应。这些结果提示,TRIM31介导了AngII引起的小鼠HRD的病理损害。课题组深入的机制研究证实,TRIM31可负向调控 TGF-β1 介导的经典 Smad 信号通路和MAPKs/NF-kB 信号通路,其作用机制是直接降解蛋白激酶 MAP3K7。总之,该研究揭示了HRD发生和发展的新机制:HRD肾脏中TGF-β1表达升高,抑制 TRIM31 的表达,进而减少 MAP3K7 的蛋白降解,通过过度激活的 Smad 信号通路和 MAPK/NF-kB信号通路,促进肾脏的纤维化及炎症反应。这些研究结果为 HRD 的治疗提供了新的靶点。
该研究发表于Nature子刊《细胞死亡和分化》(Cell Death and Differentiation,中科院一区,IF:15.828),论文的第一作者是山东大学齐鲁医院心内科博士后张杰,山东大学齐鲁医院张猛教授、张运院士和山东大学基础医学院高成江教授为该论文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。
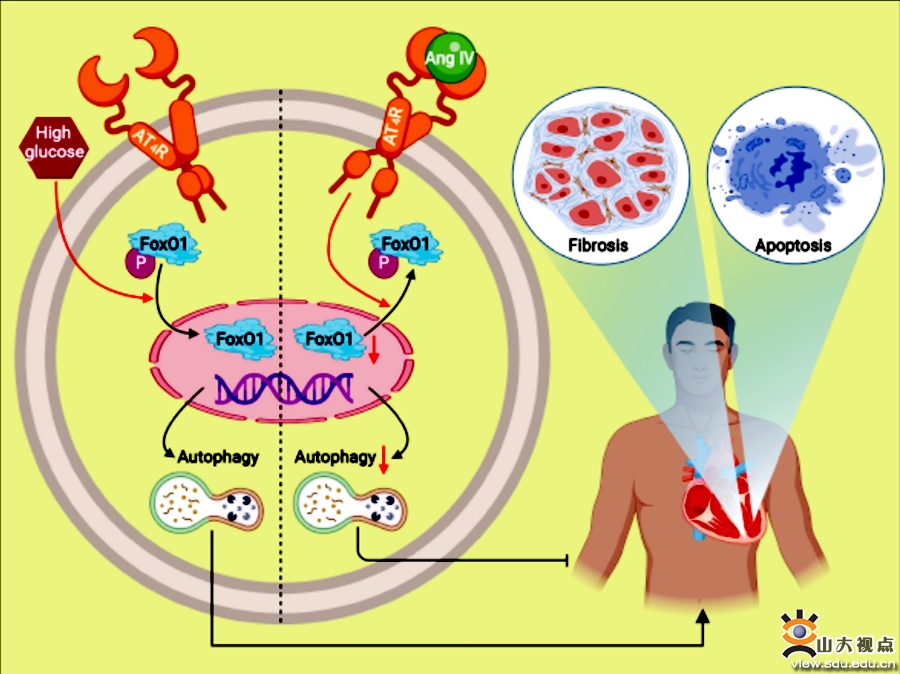
在世界范围内,糖尿病的发病率持续上升,且具有明显的年轻化趋势。糖尿病患者的糖脂代谢异常可引起心肌细胞的进行性损伤并激活心肌成纤维细胞,导致心肌间质纤维化和心脏功能的进行性减退。以往研究发现,肾素-血管紧张素系统(RAAS)过度激活和心肌细胞自噬异常可能是糖尿病导致心肌损伤和心室重构的重要机制,因此,寻找RAAS和心肌细胞自噬共同的调控因子或药物可能为改善糖尿病心肌损伤和心室重构提供重要的干预靶点。课题组首先在C57BL/6J背景的小鼠中诱发糖尿病,分别给予小鼠低、中、高三种剂量的RAAS新成员血管紧张素IV(Ang IV)干预,结果显示,未干预糖尿病小鼠心功能明显异常,而给予Ang IV干预可剂量依赖性地减轻糖尿病小鼠的心脏功能障碍。病理学研究显示,未经干预的糖尿病小鼠表现明显的心肌结构异常,经Ang IV尤其是高剂量Ang IV干预后,这些心肌形态和超微结构的异常在很大程度上得到了逆转。此外,与正常小鼠相比,糖尿病小鼠心肌中有显著的胶原沉积,I型和III型胶原蛋白以及TGF-β1的蛋白表达显著上调,心肌凋亡标志物表达明显增加,而Ang IV干预可剂量依赖性地降低这些分子的异常表达。这些结果表明,Ang IV可剂量依赖性地减轻糖尿病小鼠的心功能障碍、间质纤维化和心肌细胞凋亡。差异表达基因分析显示,糖尿病促进小鼠心肌自噬及FoxO1表达,该作用可被Ang IV剂量依赖性地抑制,而Ang IV受体AT4R拮抗剂Divalinal可完全拮抗Ang IV的心肌保护作用。与Ang IV相似,FoxO1 抑制剂AS1842856可延缓小鼠的糖尿病性心肌病的发生和发展。体外实验证明,在高糖刺激下,FoxO1可能是Ang IV-AT4R轴的下游分子。该研究结果表明,糖尿病早期给予Ang IV干预可延缓心室重构和心功能减退,并改善自噬异常。Ang IV激活AT4R后可阻断FoxO1核转位介导的心肌细胞自噬、凋亡和胶原分泌。因此,Ang IV-AT4R是心室重构和心力衰竭的潜在干预靶点,鉴于Ang IV是一个小分子多肽,因此该研究结果具有较好的转化应用价值。
该研究发表于国际期刊《治疗诊断学》(Theranostics,中科院一区,最新IF 11.556),论文的第一作者是山东大学齐鲁医院心内科章萌医师,山东大学齐鲁医院心内科张运院士、张澄教授和郝盼盼教授为该论文的共同通讯作者。
文章链接:
1.Deficient Chaperone-Mediated Autophagy Promotes Inflammation and Atherosclerosis
2.The E3 ubiquitin ligase TRIM31 plays a critical role in hypertensive nephropathy by promoting proteasomal degradation of MAP3K7 in the TGF-β1 signaling pathway
3.Angiotensin IV attenuates diabetic cardiomyopathy via suppressing FoxO1-induced excessive autophagy, apoptosis and fibrosis