近日,齐鲁医院络病理论创新转化全国重点实验室研究团队在心血管疾病基础研究领域中,发现了动脉粥样硬化(AS)斑块进展和心脏瓣膜钙化的发病新机制和干预新靶点,论文发表在Nature Communications(中科院1区Top期刊,5年IF:16.1)、Cell Death Differ(中科院1区Top期刊,5年IF:13.7)和Theranostics(中科院1区Top期刊,五年IF:12.0)。
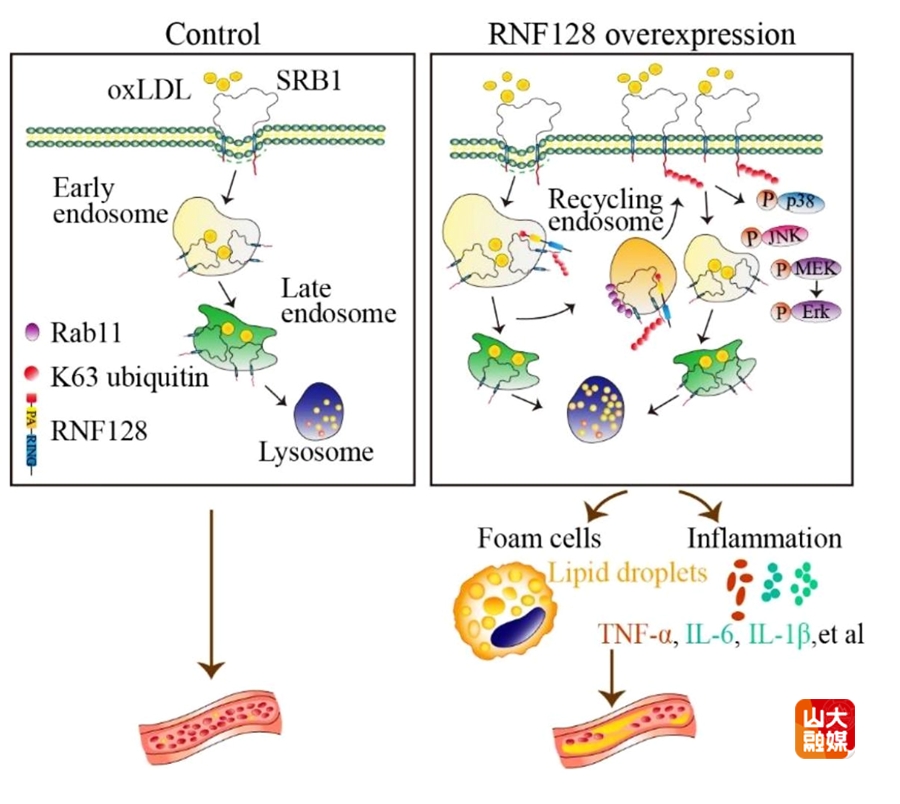
AS是动脉壁脂质沉积和慢性炎症共同作用的结果,而动脉内膜下泡沫细胞的出现是AS早期病变的标志。巨噬细胞清道夫受体主要包括清道夫受体A1(SRA1)、清道夫受体B1(SRB1)及CD36,负责识别、结合和摄取低密度脂蛋白胆固醇(LDL-C),脂质的过多摄取或外排不足都将导致泡沫细胞的形成。以往研究显示,E3泛素连接酶RNF128通过靶向结合多种膜蛋白以调控疾病的发展,但RNF128在AS中的作用和机制尚不明了。齐鲁医院全国重点实验室教授张澄、张猛和基础医学院教授高成江团队合作研究,发现E3泛素连接酶RNF128在AS斑块中的巨噬细胞亚群特异性表达,并随人类和小鼠斑块的进展表达逐渐上调。课题组构建了ApoE-/-和LDLR-/-两种AS小鼠模型以及巨噬细胞RNF128特异性敲除小鼠模型,发现巨噬细胞RNF128特异性敲除明显减轻了AS小鼠斑块的进展。在机制研究方面,课题组发现巨噬细胞RNF128敲除可减少oxLDL的内吞而抑制泡沫细胞的形成,而使用RNF128的过表达腺病毒则证明,RNF128对脂质内吞的调节依赖于RING结构域的酶活性。深入研究发现,巨噬细胞中RNF128的PA结构域可特异性靶向清道夫受体SRB1胞外结构域,通过RING结构域对SRB1的第478位赖氨酸发生K63位泛素化修饰,促使SRB1在Rab11的介导下循环至细胞膜而避免其进入溶酶体进行降解,从而促进oxLDL的过量内吞和泡沫细胞的形成。综上所述,E3泛素连接酶RNF128通过促进巨噬细胞泡沫化推动了AS的发生和发展。因此,靶向抑制巨噬细胞RNF128可作为AS的潜在治疗靶点。该研究发表于Nature Communications,张澄教授、张猛教授、高成江教授为该文的共同通讯作者,山东大学齐鲁医院刘亚鹏为该文的第一作者,山东大学齐鲁医院为该文的第一和通讯作者单位。
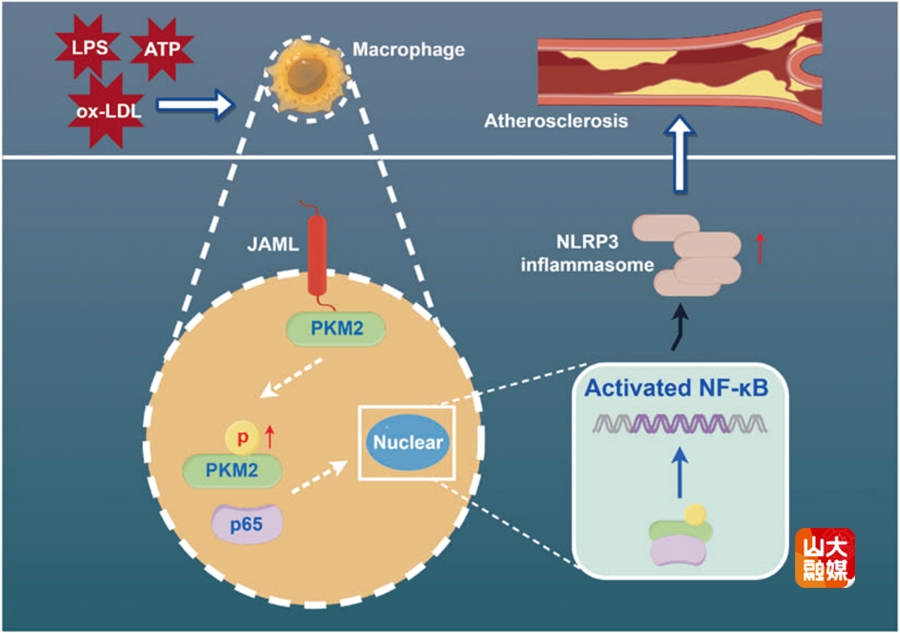
近年来研究证明,在AS发生和发展过程中,巨噬细胞中NOD样受体家族核苷酸结合寡聚化结构域样受体3(NLRP3)炎症小体起到了关键作用,激活NLRP3可促进AS进展的关键促炎因子—白细胞介素-1β(IL-1β)的分泌,然而NLRP3的调控机制仍不明确。
连接粘附分子样蛋白(JAML)是一种I型跨膜糖蛋白,介导细胞间的相互作用并与细胞内蛋白结合,进而激活下游信号通路。然而,巨噬细胞JAML在NLRP3炎症小体激活和AS发病中的作用尚不明了。齐鲁医院全国重点实验室教授杨建民、张文程团队首先分析了以往人颈动脉粥样硬化数据集,发现人AS化斑块中JAML表达的显著上调。继之,课题组构建了AS小鼠模型,发现小鼠斑块中JAML的表达显著增加,尤其在巨噬细胞中高表达。进一步研究发现,JAML在冠心病患者的人外周血单核细胞(PBMCs)和人单核细胞源性巨噬细胞(HMDMs)中的表达均显著升高。为了明确JAML在AS发生和发展中的作用,课题组构建了巨噬细胞特异性敲除和过表达JAML合并AS的小鼠模型。结果显示,巨噬细胞特异性敲除JAML可抑制小鼠AS的发展,反之,过表达JAML可促进AS的进展。深入的机制研究显示,JAML通过调节PKM2的磷酸化和核移位激活NF-κB信号通路,导致NLRP3炎症小体激活,刺激促炎因子分泌,最终加速AS的进展。这些发现首次揭示了巨噬细胞JAML在AS中的重要作用,为AS的抗炎治疗提供了潜在的新靶点。该研究发表于Cell Death and Differentiation,杨建民教授、张文程教授为该文的共同通讯作者,山东大学齐鲁医院博士研究生崔会良、博士后谢琳和博士后吕翰林为该文的共同第一作者,山东大学齐鲁医院为该文的第一和通讯作者单位。
随着全球人口老龄化趋势发展,钙化性主动脉瓣疾病(CAVD)的患病率显著增加,已成为最常见的获得性心脏瓣膜病。经导管主动脉瓣置换术作为治疗严重主动脉瓣狭窄的微创介入方案,已在临床实践中确立其治疗地位,但仍面临生物瓣膜材料的耐久性问题。因此,探索可预防、延缓或逆转CAVD进展的干预新靶点和新机制,具有重要的临床意义。

山东大学齐鲁医院全国重点实验室教授安贵鹏、张澄团队研究发现,在人和小鼠钙化主动脉瓣膜组织中AMBP表达明显升高。体外实验证实,当原代瓣膜间质细胞(VIC)暴露于钙化诱导培养基(OM)构建的病理微环境时,AMBP表达水平呈显著上调趋势。继之,课题组开展了功能实验,发现在高胆固醇饮食(HCD)诱导的ApoE-/-小鼠主动脉瓣钙化模型中,AMBP过表达显著降低了小鼠的瓣膜厚度、纤维化程度和钙化面积,改善了主动脉瓣血流动力学指标。体外实验进一步证实,AMBP过表达可显著降低VIC中OM诱导的钙化标志物(RUNX2、OSTERIX)的表达并减少钙结节沉积,而使用siRNAs靶向敲低AMBP则可逆转这一作用。为了阐明AMBP抑制主动脉瓣钙化的分子机制,课题组对上调的差异表达基因进行了KEGG及GSEA富集分析,发现MAPK通路在CAVD中显著激活。体外实验显示,OM暴露显著诱导了VIC中MAPK通路关键蛋白—ERK1/2和JNK磷酸化,敲低AMBP可进一步加剧二者的磷酸化。课题组采用泛素-蛋白酶体抑制实验、分子互作模拟、免疫共沉淀,截短体突变及MAPK通路抑制等实验方法,发现AMBP可竞争性结合FHL3的锌指结构域,这种互作阻断了FHL3对P-ERK1/2和P-JNK的保护作用,进而促进P-ERK1/2和P-JNK泛素-蛋白酶体降解。体外实验发现,ERK/JNK抑制剂可逆转AMBP敲低诱导的VIC成骨分化。最后,课题组通过体内实验发现,使用ERK激动剂和JNK激动剂预处理可部分抵消AMBP过表达对小鼠瓣膜钙化的保护作用,证实AMBP通过MAPK通路而发挥作用。综上所述,该研究首次发现AMBP调控CAVD的重要作用和分子机制,为CAVD的防治提供了新的思路和靶点。该研究发表于Theranostics杂志,安贵鹏教授、张澄教授为本文的共同通讯作者,山东大学齐鲁医院博士后郭成虎、副教授刘晓玲为该文的共同第一作者,山东大学齐鲁医院为该文的第一和通讯作者单位。