近日,齐鲁医院络病理论创新转化全国重点实验室张澄教授、张猛教授、张运院士、卜培莉教授、隋文海副教授和提蕴副教授研究团队在心血管疾病基础研究领域中,发现了动脉粥样硬化(AS)斑块进展、心肌缺血再灌注(I/R)损伤、压力超负荷心肌重构和糖尿病性肾病(DKD)的系列发病新机制和干预新靶点,论文发表在Nature旗下Signal Transduction and Targeted Therapy(中科院1区Top期刊,5年IF:52.2)、Circulation Research(中科院1区Top期刊,5年IF: 20.8)、TheJournal of Clinical Investigation(中科院1区Top期刊,5年IF:14.4)和Pharmacological Research(中科院1区Top期刊,5年IF:10.3)。
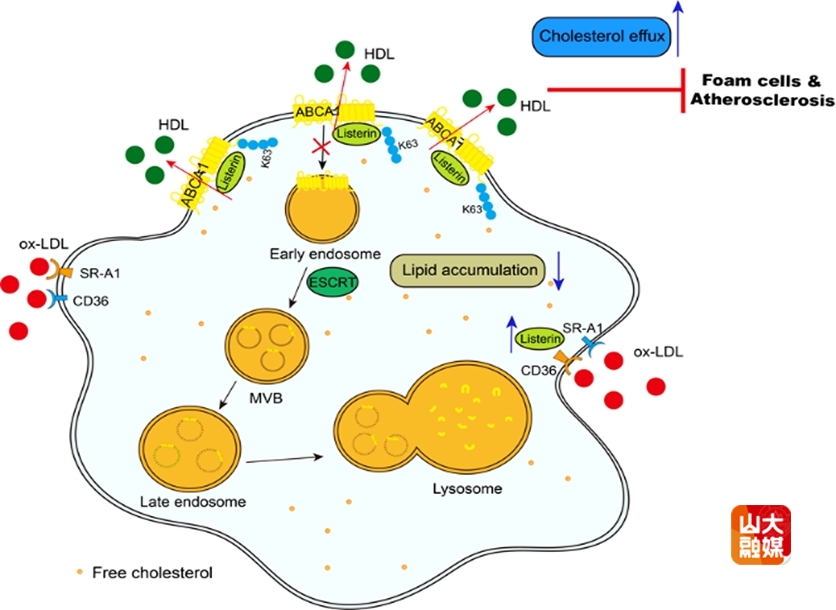
动脉粥样硬化性心血管疾病(ASCVD)是全球范围内导致疾病和死亡的首要原因。动脉粥样硬化的特点是动脉壁内膜中脂质过度沉积,而巨噬细胞吞噬脂质形成泡沫细胞在此过程中发挥着关键作用。深入研究巨噬细胞脂质代谢调控机制可为AS的防治提供新的战略视角。
以往研究显示,在酵母和细菌中,E3泛素连接酶Listerin的同源类似物LTN1可作为核糖体相关蛋白质质量控制系统的组成部分,介导新生肽链的泛素化修饰和降解。然而,哺乳动物Listerin调控AS的机制仍不明了。张澄教授和张猛教授以及山东大学基础医学院高成江教授课题组研究发现,Listerin表达与AS的进展存在正相关,巨噬细胞特异性敲除Listerin可显著加速AS斑块进展和斑块脂质沉积。课题组利用蛋白组学测序发现,Listerin敲除后胆固醇外排关键作用蛋白ABCA1表达明显降低,小鼠AS模型研究证实,Listerin以ABCA1依赖方式调控AS的进展。深入的机制研究发现,Listerin可通过K63位的泛素化修饰抑制ABCA1通过ESCRT溶酶体途径的降解,进而促进巨噬细胞胆固醇外排并减缓泡沫化进程。该发现为AS的防治提供了崭新的靶点。该研究发表于国际权威期刊The Journal of Clinical Investigation,山东大学齐鲁医院心血管内科博士后曹磊、张杰和俞力雯是该文的共同第一作者,张澄教授、张猛教授、高成江教授为该文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。
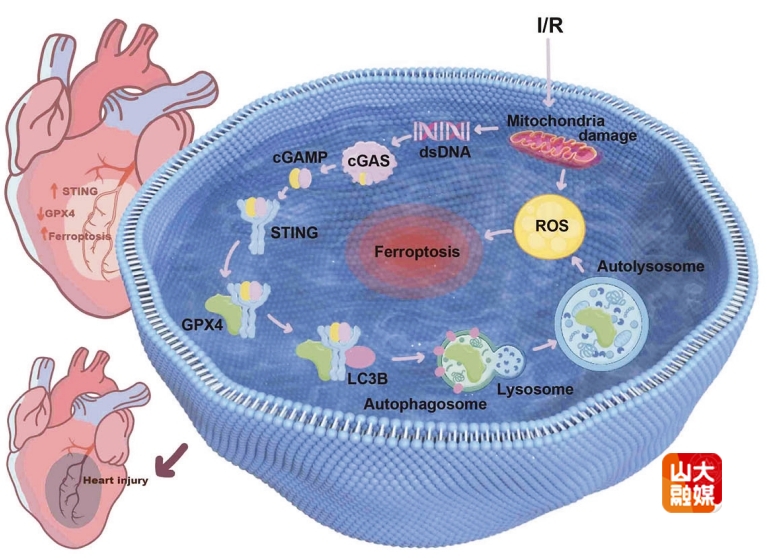
缺血性心脏病是全球主要的致死原因之一。尽管心肌梗死后的介入技术已取得显著进展,但仍有相当一部分患者因心肌I/R损伤而面临较高的死亡风险。深入探究I/R损伤的发生机制,对于保护缺血心肌、增加患者生存至关重要。
干扰素基因刺激因子STING定位于内质网,是抗病毒天然免疫及肿瘤免疫信号转导中的关键节点蛋白。细胞内核酸受体cGAS感知外源性病毒DNA或细胞应激状态下释放的dsDNA后,产生第二信使cGAMP,诱导STING激活,启动下游TBK1-IRF3信号的级联反应,产生I型干扰素、炎症因子等发挥免疫调节作用。近年研究显示,cGAS-STING激活后可通过不依赖于TBK1-IRF3的非经典途径发挥重要作用,探寻STING的新机制和新靶点是当前国际研究的前沿。张澄教授和张猛教授课题组首次发现,在心肌缺血再灌注过程中,胞质双链DNA积累,激活cGAS-cGAMP-STING信号通路,激活的STING直接结合铁死亡关键调控蛋白GPX4并诱导其自噬性降解。以上过程引发进一步的氧化应激,最终触发心肌细胞的铁死亡,加剧心肌I/R损伤。相反,应用STING小分子抑制剂H-151可有效抑制GPX4的降解,显著改善心肌缺血后的心脏损伤。该项研究首次揭示了STING在心肌细胞中启动铁死亡并加重心肌I/R损伤的机制,为缺血性心脏病的治疗提供了新的干预靶点。该研究发表于国际权威期刊Signal Transduction and Targeted Therapy,齐鲁医院心血管内科博士研究生王晓宏为该文的第一作者,张澄教授和张猛教授为该文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。
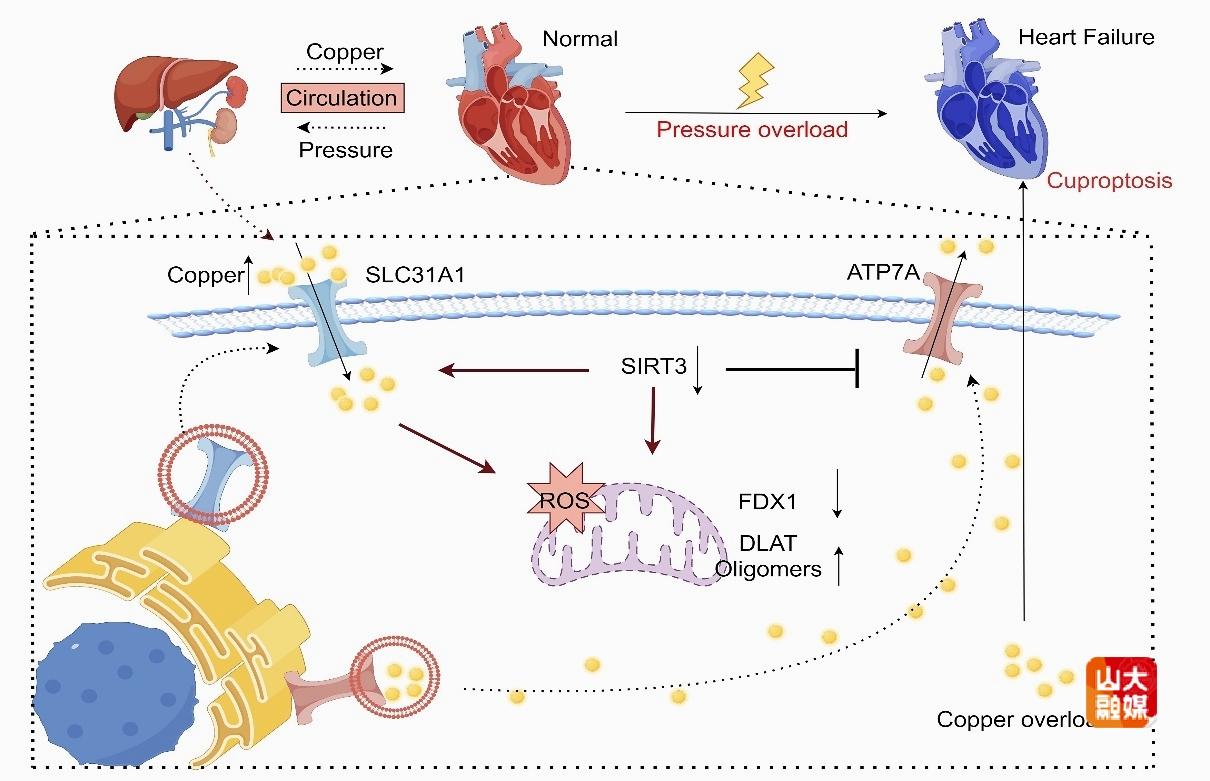
病理性心肌重构是压力超负荷诱导心力衰竭的关键环节,铜死亡作为由铜离子引发的细胞新型死亡方式,在心肌重构过程中的作用尚不明确,深入探讨心肌重构的发生机制,将为开发更为有效的预防与治疗策略提供重要科学依据。
Sirtuin3(SIRT3)是定位于线粒体的去乙酰化酶,对于调控能量代谢和氧化应激起有重要作用。卜培莉教授团队发现,心力衰竭患者中SIRT3相关的铜死亡差异基因集中于铜离子转运蛋白簇,在压力超负荷诱导的心肌重构模型中心肌组织SIRT3表达下调,心肌细胞表现出铜死亡特征。据此,课题组构建了SIRT3基因敲除与过表达小鼠模型,发现SIRT3可有效改善压力超负荷诱导的心肌重构,减少心肌组织内铜离子积累与细胞铜死亡。 机制研究发现,铜离子转运蛋白ATP7A和SLC31A1均具有LIR基序,通过分子模拟对接、免疫共沉淀、自噬流干扰等方法证明,SIRT3可通过调节高铜刺激下的细胞自噬水平,影响铜离子转运蛋白与自噬标志蛋白LC3B II的结合,改变细胞内铜离子转运蛋白的自噬途径降解。本研究揭示了SIRT3通过自噬调节铜离子转运蛋白含量,维持高铜环境下细胞内铜离子稳态,进而减轻心肌细胞铜死亡的新机制,为压力超负荷诱导的病理性心肌重构提供了潜在的干预靶点。该研究发表于国际权威期刊Pharmacological Research,齐鲁医院心血管内科博士研究生孔炳辉为该文的第一作者,卜培莉教授和提蕴副教授为本文的共同通讯作者,山东大学齐鲁医院为第一和通讯作者单位。
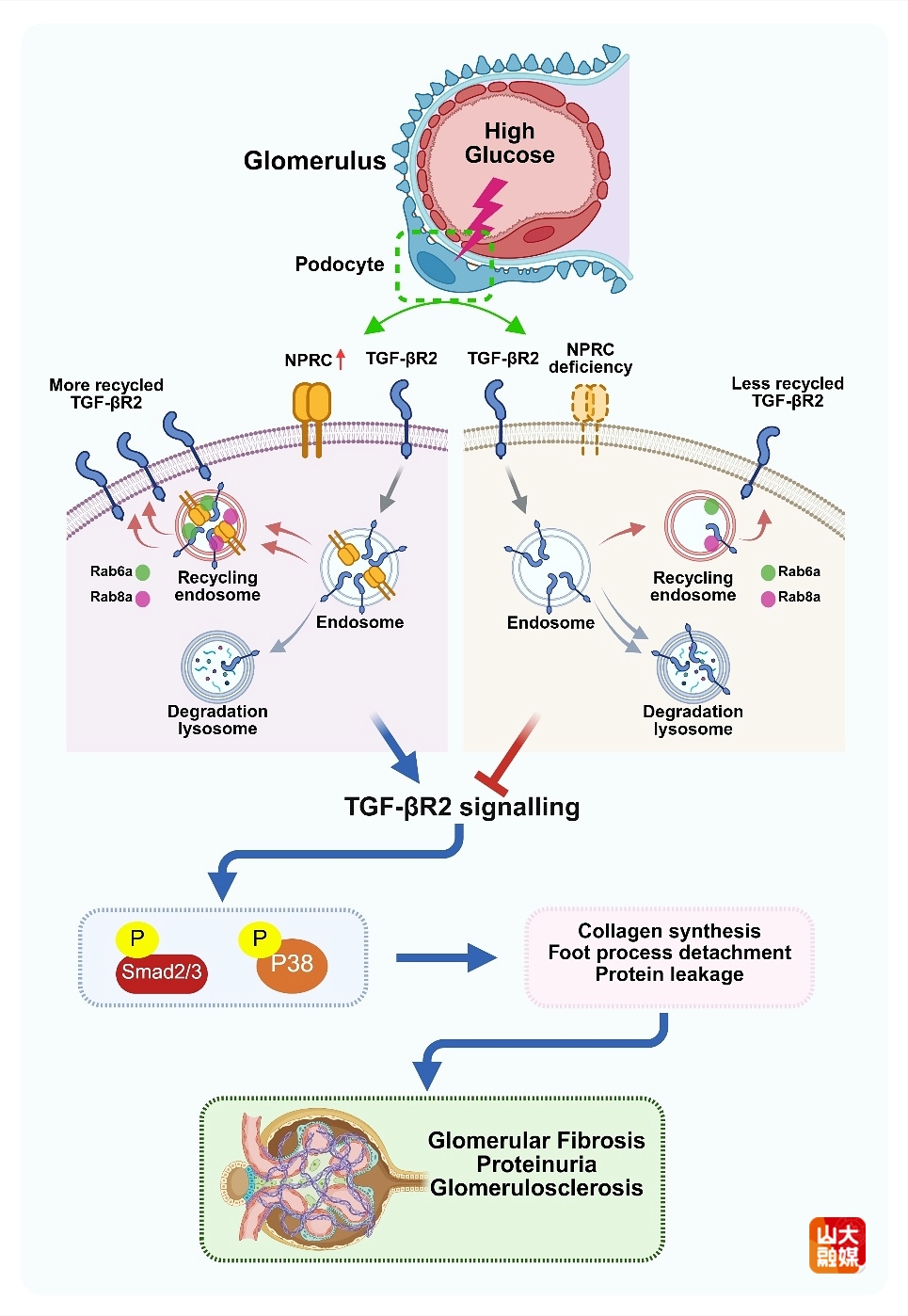
近十年来,我国糖尿病的发病率迅速上升,糖尿病肾病(DKD)是糖尿病最常见和严重的并发症之一,常导致肾功能衰竭。肾小球的足细胞损伤在DKD的早期阶段起有重要作用,且与DKD的严重程度相关。然而,临床仍缺乏干预DKD的有效手段,探索DKD的发病机制和干预靶点具有重要的临床意义。
利钠肽(NPs)是一组参与调节肾脏、心血管、内分泌和骨骼稳态的肽类激素。利钠肽受体C(NPRC)是一种与三种NPs都具有高亲和力的受体。NPRC的主要功能是通过激活抑制性鸟苷调节蛋白和抑制环腺苷酸(cAMP),增强溶酶体降解途径,进而清除循环中过量的NPs。NPRC在肾脏中的分布密度远高于在其他组织,但对DKD的影响及其机制尚不明了。张澄教授、张运院士和隋文海副教授课题组首次发现,在DKD患者和小鼠肾脏中NPRC表达增加,且以往DKD小鼠肾脏单细胞测序数据分析表明,NPRC主要在足细胞中表达。继之,课题组构建了足细胞特异性NPRC基因敲除小鼠,结果显示NPRC基因敲除可改善DKD小鼠血清肌酐和尿白蛋白水平、抑制肾脏增大、减轻系膜基质堆积和肾小球基底膜增厚、改善足细胞损伤。深入的机制研究发现,足细胞中NPRC的缺乏可抑制TGF-βR2再循环后的重新上膜、促进其进入降解途径,从而减少TGF-βR2的蛋白水平,进而抑制TGF-β1/Smad和TGF-β1/P38信号通路,减少肾小球纤维化并改善肾功能。这些发现为DKD的防治提供了潜在的治疗新靶点。该研究发表于国际权威期刊Circulation Research,齐鲁医院心血管内科博士研究生王鑫陆是该文的第一作者,张澄教授、张运院士和隋文海副教授为该文的共同通讯作者。山东大学齐鲁医院为第一和通讯作者单位。